上线-Meta打造首个「蛋白质宇宙」全景图!用150亿参数语言模型,预测了6亿+蛋白质结构

新智元报道
编辑:好困 David
【新智元导读】Meta AI在蛋白质结构预测上又有新突破!利用大型语言模型 ,打造了一个超过6亿个宏基因组结构的数据库,让蛋白质结构预测提速60倍。
Meta在蛋白质结构的探索上又前进了一步!
这次他们瞄准的是更大的目标领域:宏基因组学。

蛋白质宇宙的「暗物质」
根据NIH人类基因组研究所的解释,宏基因组学(Metagenomics,又译宏基因组学)研究从大宗样品中的所有生物体(通常是微生物)中分离和分析的整个核苷酸序列的结构和功能,通常用于研究特定的微生物群落,比如居住在人类皮肤上、土壤中或水样中的微生物身上的蛋白质。
在过去的几十年里,随着我们对生活在人类体内、身上以及环境中的所有微生物有更多的了解,宏基因组学一直是一个非常活跃的领域。
由于宏基因组学的研究对象无所不包,远远超过了构成动植物生命的蛋白质,可以说是地球上最不为人知的蛋白质。
为此,Meta AI用上了最新的大型语言模型、打造了一个超过6亿个宏基因组结构的数据库,并提供一个API,让科学家轻松检索与工作相关的特定蛋白质结构。

论文地址:https://www.biorxiv.org/content/10.1101/2022.07.20.500902v2
Meta表示,解码宏基因组结构,有助于解开长期存在的人类进化史之谜,帮助人类更有效地治愈疾病、净化环境。
蛋白质结构预测,提速60倍!
宏基因组学主要研究如何从所有这些在环境中共存的生物体中获得DNA,这有点像一盒拼图,但并不只是一盒拼图,实际上是所有10组较小的拼图堆在一起,放在一个盒子里。
宏基因组学同时获取这10种生物的基因组时,实际上是试图同时解决10个谜题,了解同一个基因组盒子里的所有不同的拼图。
正是这种结构和生物作用的未知性,通过宏基因组学发现的新蛋白质,甚至可以称为蛋白质宇宙的「暗物质」。

近些年,基因测序方面的进步让编目数十亿宏基因组蛋白质序列成为可能。
然而,尽管已经知道这些蛋白质序列的存在,但想要进一步了解它们的生物学特性,却是一个巨大的挑战。
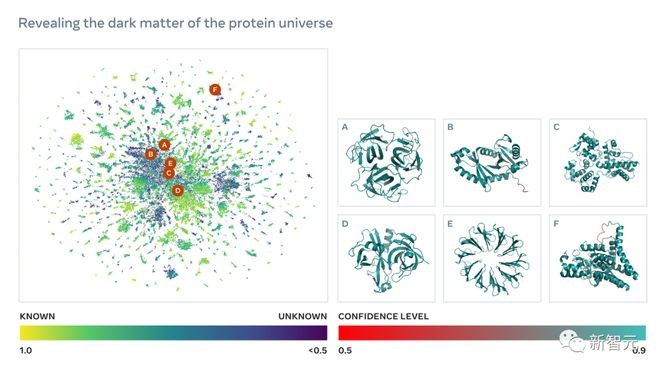
为了要得到这些数以亿计的蛋白质序列结构,预测速度的突破是至关重要的。
这个过程,即便是用目前最先进的工具,再搭上一个大型研究机构的计算资源,也可能需要数年时间。
于是,Meta训练了一个大型语言模型,来学习进化模式,并直接从蛋白质序列中端到端地生成准确的结构预测,在保持准确性的同时,预测速度比当前最先进的方法快60倍。
事实上,借助于这种新的结构预测能力,Meta在短短两周内用一个由大约2000个GPU组成的集群上,预测出了图谱中超过6亿个宏基因组蛋白质的序列。

Meta发布的宏基因组图谱名为ESM Atlas,几乎涵盖了整个宏基因组序列公共数据库MGnify90的预测。
Meta表示,ESM Atlas是迄今为止最大的高分辨率预测结构数据库,比现有蛋白质结构数据库大3倍,而且是第一个全面和大规模地覆盖宏基因组蛋白质的数据库。
这些蛋白质结构为了解自然界的广泛性和多样性提供了一个前所未有的视角,并有可能加速发现蛋白质在医学、绿色化学、环境应用和可再生能源等领域的实际应用。
此次用于预测蛋白质结构的新语言模型拥有150亿个参数,是迄今为止最大的「蛋白质语言模型」。
这个模型实际上是Meta今年7月发布的ESM Fold蛋白质预测模型的延续。
在当初ESMFold发布时,已经和AlphaFold2、RoseTTAFold等主流蛋白质模型不相上下。但ESMFold预测速度要比AlphaFold2快一个数量级!
一下说数量级可能不好理解三者之间速度的对比,看看下面这张图就懂了。
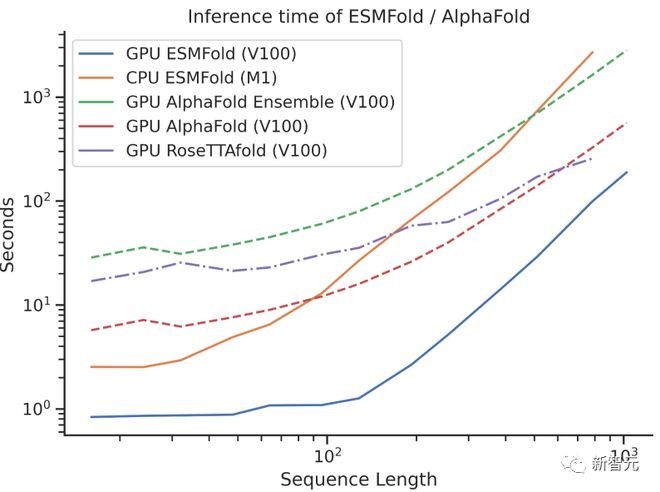
而这次ESM Atlas数据库的发布,更是让150亿参数的大语言模型有了最广泛的用武之地。
由此,科学家便可以在数亿个蛋白质的规模上搜索和分析以前没有被定性的结构,并发现在医学和其他应用中有用的新蛋白质。
语言模型,真是「万能」的
就像文本一样,蛋白质也可以被写成字符序列。
其中,构成蛋白质的每个「字符」都对应于20个标准化学元素之一——氨基酸。而每个氨基酸又都有不同的特性。
但想要读懂这种「生物学语言」是个很大的挑战。
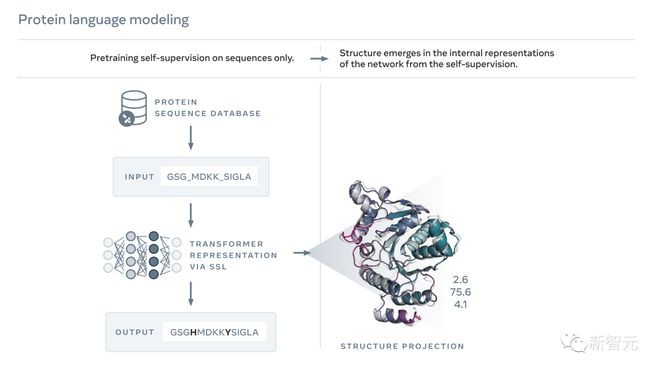
虽然就像刚刚说的,蛋白质序列和一段文字都可以写成字符,但它们之间却存在着深刻而根本的区别。
一方面, 这些 「 字符」 的不同组合方式的数量是个天文数字。 例如,对于由200个氨基酸组成的蛋白质,有20^200 种可能的序列,比目前可探索的宇宙中的原子数量还要多。
另一方面,氨基酸的每个序列都会根据物理定律折叠成一个三维形状。而且,不是所有的序列都会折叠成连贯的结构,其中有很多会折叠成无序的形式,但正是这种让人捉摸不透形状决定了蛋白质的功能。
举个例子,如果一个位置出现了某种氨基酸,而这种氨基酸通常又会与另一个位置的某种氨基酸配对。那么 ,在之后的折叠结构中,它们就很可能存在 相互作用。
而人工智能,便可以通过观察蛋白质序列来可以学习和阅读这些模式,进而推断出蛋白质的实际结构。
在2019年,Meta 提出了语言模型学习蛋白质属性的证据,例如它们的结构和功能。

论文地址:https://www.pnas.org/doi/10.1073/pnas.2016239118
利用掩膜这种自监督学习形式训练的模型,可以正确地填补一段文字中的空白,如「要不要__,这是________」。
通过这 种方法, Meta 在数百万天然蛋白质序列的基础上上训练了一个语言模型,从而实现了填补 蛋白质序列中的空白,如「GL_KKE_AHY_G」。
实验表明,这种模型经过训练,可以发现关于蛋白质的结构和功能的信息。
2020年,Meta发布ESH1b,这是当时最先进的蛋白质语言模型,目前已被用于各种应用,包括帮助科学家预测新冠病毒的进化,以及发现遗传病的病因。

论文地址:https://www.biorxiv.org/content/10.1101/2022.08.25.505311v1
现在,Meta扩大了这种方法的规模,创建了下一代蛋白质语言模型ESM-2,这是一个150亿参数的大模型。
随着模型从800万个参数扩展到1500万个参数,内部表征中出现的信息能够在原子分辨率下进行三维结构预测。
读懂「蛋白质语言」,让生命更透明
从几十亿年前起,生物的进化就形成了一种蛋白质语言,这种语言可以通过简单的构件形成复杂而动态的分子机器。学习阅读蛋白质的语言是我们理解自然界的一个重要步骤。
AI可以为我们提供理解自然世界的新工具,就像显微镜一样,让我们以几乎无限小的尺度来观察世界,并开启了对生命的全新理解。AI可以帮助我们理解自然界多样性的巨大范围,并以一种新的方式看待生物学。
目前,大部分的AI研究都是让计算机以类似于人类的方式来理解世界。蛋白质的语言是人类无法理解的,甚至最强大的计算工具也无法理解。
所以,Meta的这项工作的意义在于揭示了AI在跨领域时的巨大优势,即:在机器翻译、自然语言理解、语音识别和图像生成方面取得进展的大型语言模型,也能够学习有关生物学的深刻信息。
此次Meta公开这项工作,分享数据和成果,并以他人的见解为基础,希望这个大规模结构图集和快速蛋白质折叠模型的发布,可以推动进一步的科学进步,使我们更好地了解周围的世界。
参考资料:
https://ai.facebook.com/blog/protein-folding-esmfold-metagenomics/?utm_source=twitter&utm_medium=organic_social&utm_campaign=blog